
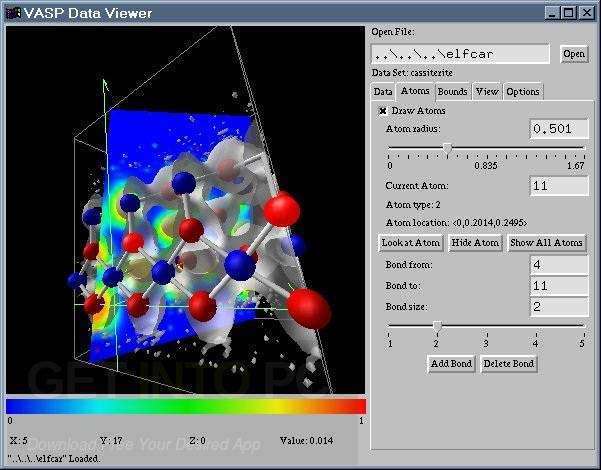
Download VASP 6.5.1 – Comprehensive Quantum Mechanical Simulation Software
The Vienna Ab initio Simulation Package (VASP) is a sophisticated computational chemistry tool developed by the VASP group at the University of Vienna. This software package is designed for atomic-scale materials modeling, employing quantum mechanical principles to simulate molecular dynamics for systems such as solids and molecules. VASP is instrumental in material science, quantum chemistry, physics, and engineering, offering users the capability to perform detailed electronic structure calculations and analyze reaction pathways.
Introduction to VASP
VASP stands as a fundamental tool in computational chemistry and material science, offering advanced capabilities for simulating atomic-scale phenomena. Its application of quantum mechanical methods, particularly density functional theory, allows for precise investigations into the behavior of materials at the molecular level. The software enables researchers to delve into electronic structures, optimize geometries, and understand complex material properties, making it an indispensable resource for scientific discovery and engineering applications.
Key Features and Functionalities of VASP
Dynamics and Relaxation Techniques
VASP provides a range of advanced techniques for simulating molecular dynamics and optimizing material structures. These methods are crucial for understanding the behavior of systems under various conditions and for finding stable configurations.
- Oppenheimer Molecular Dynamics: Facilitates simulations that directly couple electronic structure calculations with ionic motion, enabling accurate modeling of dynamic processes.
- Force Calculations: Supports precise calculation of forces acting on atoms, which is essential for geometry optimization and molecular dynamics.
- Transition State Search: Offers functionalities to locate transition states in chemical reactions, providing insights into reaction mechanisms and activation energies.
Electronic Structure Calculations
The core of VASP’s capability lies in its robust electronic structure calculations, which reveal detailed information about a material’s bonding and electronic properties. The efficient handling of pseudopotentials is a key aspect of its performance.
- Pseudopotential Methods: VASP utilizes pseudopotentials to simplify calculations by treating core electrons and nuclei as a single unit, significantly improving computational efficiency and accuracy for valence electron interactions.
- Density Functional Theory (DFT): Implements various DFT functionals to approximate the exchange-correlation energy, forming the basis for many electronic structure studies.
- Band Structure Calculations: Enables the computation of electronic band structures, critical for understanding a material’s conductivity and optical properties.
Applications in Material Science and Chemistry
VASP’s versatility makes it applicable across a wide spectrum of scientific disciplines, from fundamental research to practical engineering challenges.
- Solid-State Reactions: Simulating chemical reactions and phase transformations within solid materials.
- Nanotechnology: Modeling the properties and behaviors of nanomaterials, such as quantum dots and nanotubes.
- Materials Engineering: Designing and predicting the properties of new alloys, ceramics, and polymers for specific applications.
- Catalysis Research: Investigating reaction pathways and mechanisms on catalytic surfaces.
- Surface Science: Studying adsorption, diffusion, and reactions on material surfaces.
Advanced Functionalities
Response to External Fields
VASP extends its analytical capabilities to include the simulation of how materials respond to external electric and magnetic fields, providing deeper insights into their physical characteristics.
- Dielectric Properties: Calculates static and frequency-dependent dielectric tensors, essential for understanding how materials interact with electric fields.
- Piezoelectric Response: Detects and quantifies piezoelectric effects, relevant for sensors and actuators.
- Magnetic Response Functions: Computes various magnetic susceptibility tensors to characterize a material’s behavior in magnetic fields.
Magnetic Properties and Berry Phases
The software allows for detailed analysis of magnetic properties, including the calculation of finite magnetic moments and the implications of Berry phases in electronic structures.
- Finite Magnetic Moments: Calculates and analyzes spin polarization effects, enabling the study of magnetism in materials.
- Berry Phase Calculations: Determines Berry phases, which are crucial for understanding topological properties of electronic systems, such as the quantum Hall effect.
- Spin-Orbit Coupling: Includes effects of spin-orbit interaction, which is vital for understanding magnetic anisotropy and topological insulators.
Comparative Analysis with Other Computational Tools
VASP distinguishes itself from other computational chemistry software through several key features that enhance its applicability and accuracy in materials simulation. While packages like Quantum ESPRESSO and Gaussian offer powerful quantum mechanical calculations, VASP provides a unique blend of capabilities.
VASP is particularly noted for its integration of both classical and quantum mechanics, along with efficient pseudopotential retrieval methods. This, combined with its robust handling of complex systems and advanced functions like explicit calculation of electric and magnetic field responses, positions it as a versatile tool for material science. Many users find its comprehensive nature and specific implementations for molecular dynamics and electronic structure analysis particularly beneficial for their research workflows.
Frequently Asked Questions
What is VASP used for?
VASP is used primarily for simulating atomic-scale systems, allowing researchers to study electronic structures and molecular dynamics. It’s widely relied upon in fields like material science, chemistry, and condensed matter physics for tasks ranging from predicting material properties to understanding reaction mechanisms.
How does VASP compare to other quantum simulation software?
VASP distinguishes itself with its robust handling of pseudopotential methods and integration of both classical and quantum mechanics, making it effective for various material types. Many users find its interface and support particularly beneficial compared to alternatives like Quantum ESPRESSO, especially for its advanced capabilities in response functions.
What are the latest features in VASP 6.5.1?
Version 6.5.1 introduces enhancements in algorithmic efficiency and accuracy, including improved methods for computing dielectric properties and magnetic responses, making it a powerful tool for modern computational studies. These updates aim to provide more precise and faster calculations for complex material systems.







Reviews
There are no reviews yet.