
Download BioSolveIT SeeSAR – Advanced Computational Chemistry Software
BioSolveIT SeeSAR is a leading computational chemistry software developed by BioSolveIT GmbH, specifically engineered for molecular modeling and simulation within the demanding fields of drug discovery and pharmaceutical research. This advanced application empowers scientists by facilitating the interactive prioritization of molecular candidates, thereby streamlining and enhancing the critical drug design process. Professionals in computational chemistry and molecular modeling rely on SeeSAR for its robust capabilities that bridge the gap between theoretical design and empirical validation.
Introduction to BioSolveIT SeeSAR
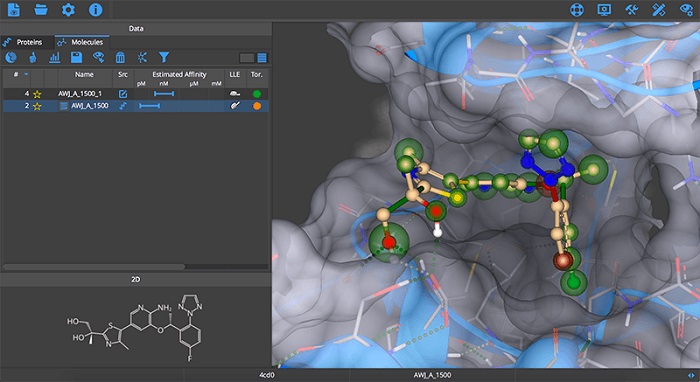
BioSolveIT SeeSAR stands as a pivotal tool in the realm of computational chemistry, offering sophisticated functionalities for molecular modeling and simulation. Its primary contribution lies in accelerating the intricate process of drug discovery, enabling researchers to analyze and optimize potential therapeutic compounds more efficiently. By providing intuitive graphical representations and robust analytical tools, SeeSAR supports decision-making in the early stages of pharmaceutical research, making it an indispensable application for scientists focused on molecular design and simulation.
Key Features of SeeSAR
Interactive Molecular Design
At the core of SeeSAR’s utility is its capacity for interactive molecular design. This feature allows researchers to conduct interactive combination prioritization, exploring various atom and molecule combinations to identify optimal structures. This iterative process is crucial for fine-tuning molecular properties during the early phases of drug development.
Rapid Parameter Optimization
SeeSAR excels in its rapid parameter optimization capabilities, a critical aspect for achieving accurate molecular simulations. The software is designed to efficiently adjust simulation parameters, ensuring that the virtual models closely mirror real-world molecular behaviors and conditions, which is essential for reliable predictive modeling.
Superior Graphic Representation
The software provides superior graphic representation tools that are vital for understanding complex molecular data. These tools offer clear and detailed visualization of atomic bonds, molecular structures, and interactions, presenting this information in an easily interpretable graphical format for enhanced analysis and comprehension.
Applications in Drug Discovery
BioSolveIT SeeSAR plays a significant role in numerous real-world drug discovery workflows. Its advanced simulation and modeling features are applied in scenarios such as lead optimization, where existing drug candidates are refined to improve efficacy and reduce side effects. Furthermore, it is instrumental in molecular docking studies, predicting how potential drug molecules will bind to target proteins.
- Lead optimization in pharmaceutical R&D.
- Precise molecular docking studies.
- Comprehensive analysis of structure-activity relationships (SAR).
- Interactive exploration of chemical compound spaces.
- Simulation of pharmacokinetic properties, including improvements in free energy calculations (ΔS/ΔH).
Comparison with Other Computational Chemistry Tools
Compared to other computational chemistry software, BioSolveIT SeeSAR distinguishes itself through its unique combination of an intuitive user interface and specialized features tailored for drug discovery. While many tools offer molecular modeling capabilities, SeeSAR’s emphasis on interactive design and its enhanced approximation of free energy calculations for pharmacokinetic modeling offer a distinct advantage for researchers focused on accelerating the drug development pipeline.
User Experience and Interface
BioSolveIT SeeSAR is recognized for its user-friendly interface, which is a significant asset for professionals working with complex computational chemistry tasks. The design prioritizes simplicity and clarity, integrating advanced functionalities within an accessible framework. This approach allows researchers to leverage powerful simulation and analysis tools without an overwhelming learning curve, thereby enhancing productivity and focus on scientific objectives.
Conclusion and Future Prospects of SeeSAR
BioSolveIT SeeSAR continues to be a powerful ally in the advancement of computational chemistry and molecular modeling. Its ongoing development, including improvements in version 114.1 for free energy calculations, demonstrates a commitment to supporting critical research areas like drug discovery. As computational methods become increasingly integral to scientific research, SeeSAR is well-positioned to further enhance the efficiency and accuracy of molecular simulation and design processes, contributing to future breakthroughs in pharmaceuticals and beyond.
Frequently Asked Questions
What is BioSolveIT SeeSAR used for?
BioSolveIT SeeSAR is primarily used in computational chemistry for molecular modeling and simulation, particularly within drug discovery processes. It allows researchers to optimize molecular combinations interactively and to perform detailed analyses essential for developing new therapeutic agents.
What are the advantages of using SeeSAR over other molecular modeling tools?
SeeSAR offers an intuitive user interface and unique features like interactive molecular design and rapid parameter optimization, which enhance the reliability of simulations and reduce the time required in the drug design process. Its specialized focus on drug discovery workflows provides advantages for researchers in this field.
How does SeeSAR improve parameter optimization for drug design?
SeeSAR improves parameter optimization through advanced algorithms that increase the accuracy of free energy calculations, ensuring that simulated samples closely represent real-world molecular interactions. This leads to more reliable predictions and better-informed design decisions.







Reviews
There are no reviews yet.