
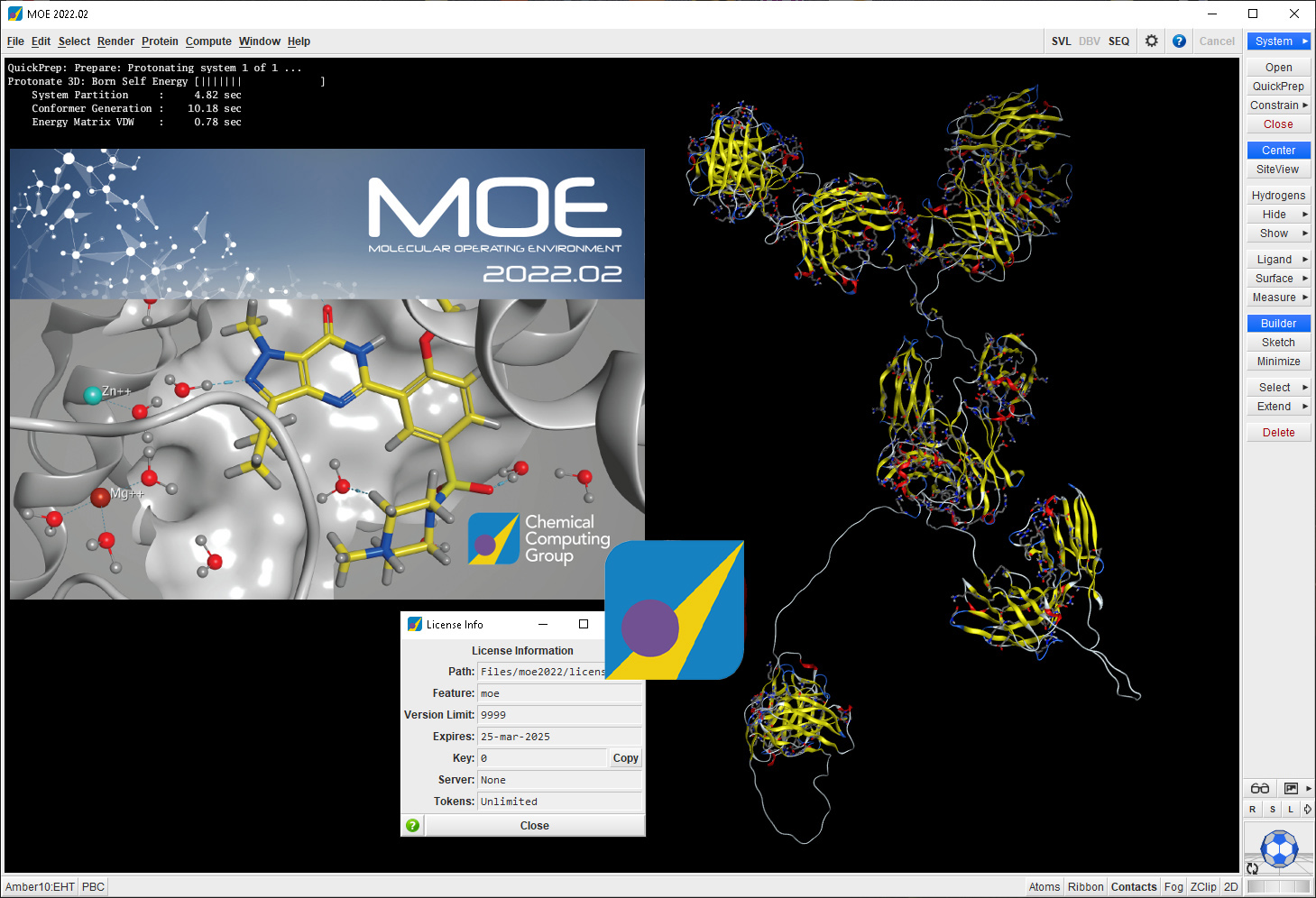
Download Chemical Computing Group MOE 2022.02 – Advanced Computational Chemistry Software
The Molecular Operating Environment (MOE) 2022.02, developed by Chemical Computing Group, is a comprehensive computational chemistry software designed for drug discovery. This platform integrates advanced visualization, modeling, and simulation tools, making it an essential application for professionals in the pharmaceutical, biotechnology, and academic research sectors. Its continuous development, guided by community input, has established MOE as a significant tool in computational chemistry.
Introduction to Molecular Operating Environment
Overview and Historical Context
Developed by Chemical Computing Group since its founding in 1994, the Molecular Operating Environment (MOE) serves as a sophisticated platform for computational chemistry and drug discovery. It offers an integrated environment that combines powerful visualization capabilities with advanced modeling and simulation tools. This holistic approach aims to streamline complex research workflows, enabling scientists to explore molecular interactions and design novel compounds more efficiently. MOE’s long history and continuous evolution highlight its sustained importance in scientific research.
Key Features of MOE 2022.02
Innovative Scientific Applications
MOE 2022.02 provides a robust suite of scientific applications tailored for deep molecular analysis and design. Its feature set is engineered to support intricate research needs within drug discovery and computational chemistry, leveraging scientific community feedback for continuous improvement.
- Protein-Ligand Design: Facilitates detailed examination and design of interactions between proteins and potential drug molecules, crucial for identifying effective therapeutic candidates.
- Bioinformatics and Cheminformatics: Offers extensive tools for managing, analyzing, and interrogating biological and chemical data, enabling comprehensive study of molecular properties and relationships.
- Molecular Simulations: Supports various simulation techniques to predict molecular behavior, stability, and reactivity under different conditions.
- Pharmacophore Modeling: Enables the identification and optimization of molecular features essential for biological activity.
- Ligand-Receptor Interactions: Provides sophisticated methods for understanding and predicting how small molecules bind to biological targets.
Supported Functionality and Integration
Operating Platforms and Computational Techniques
The Molecular Operating Environment is designed for broad accessibility and efficient computation across diverse research environments.
- Cross-Platform Compatibility: MOE 2022.02 runs seamlessly on major operating systems, including Windows, Linux, Unix, and macOS, ensuring flexibility for research teams with varied computing infrastructure.
- Scientific Vector Language (SVL): Integrates SVL, a proprietary scripting language, allowing users to automate tasks, customize workflows, and develop bespoke applications for specialized computational chemistry needs.
- Advanced Modeling Techniques: Employs a range of computational methods to simulate molecular dynamics, conformational analysis, and quantum mechanical calculations. This supports in-depth investigation of molecular structures and properties.
Applications in Drug Discovery and Biochemistry
Utilization in Pharmaceutical Research
MOE is a pivotal tool in modern drug discovery pipelines, enabling scientists to address complex challenges in identifying and optimizing therapeutic agents.
- Structure-Based Drug Design: Researchers utilize MOE to design drug candidates that precisely fit the binding sites of target proteins, a critical approach for developing highly specific and effective medications.
- Virtual Screening: The platform supports high-throughput virtual screening of compound libraries to identify potential hits that interact with specific biological targets, accelerating the initial stages of drug discovery.
- Lead Optimization: MOE assists in refining the properties of drug leads to improve efficacy, safety, and pharmacokinetic profiles through detailed analysis of molecular interactions and property predictions.
- Biomolecular Modeling: Used extensively for modeling protein structures, nucleic acids, and other biomolecules, facilitating a deeper understanding of biological mechanisms relevant to disease.
Enhancements in the 2022.02 Version
New Features and Improvements
The MOE 2022.02 release introduces several enhancements aimed at improving user experience and expanding its computational capabilities, reflecting ongoing development based on user needs and scientific advancements.
- User Interface Optimizations: The latest version includes refinements to the user interface, enhancing ease of use and accessibility for both new and experienced users.
- Advanced Search Options: Features for searching chemical databases and structural libraries have been improved, allowing for more precise and efficient data retrieval.
- Enhanced Molecular Docking: Capabilities for molecular docking have been refined, providing more accurate predictions of ligand binding poses and affinities to protein targets.
- Expanded Cheminformatics Tools: New functionalities and improvements to existing tools in cheminformatics enable more sophisticated analysis of chemical data and molecular properties.
Comparison with Other Computational Tools
MOE distinguishes itself in the computational chemistry landscape through its comprehensive integration and specialized features.
- Compared to other software packages focused solely on molecular visualization or simulation, MOE offers a unified platform that seamlessly integrates these functionalities with extensive cheminformatics and drug design tools.
- Its robust support for protein-ligand interactions and structure-based design, combined with the flexibility of the Scientific Vector Language (SVL) for customization, positions it as a strong choice for research-intensive pharmaceutical and biotechnology applications.
- While other tools might excel in specific niche areas, MOE provides a balanced and extensive toolkit suitable for a wide array of drug discovery and computational chemistry projects.
Conclusion and Future Directions
The Molecular Operating Environment (MOE) 2022.02 by Chemical Computing Group continues to serve as a cornerstone application for computational chemistry and drug discovery. Its integrated approach to visualization, modeling, and simulation, coupled with ongoing enhancements, empowers researchers in pharmaceutical, biotechnology, and academic settings to push the boundaries of molecular science. Future developments are likely to focus on further expanding its predictive capabilities and integrating emerging computational techniques to support the ever-evolving demands of modern scientific research.
Frequently Asked Questions
What is the Molecular Operating Environment (MOE) used for?
MOE is primarily used for drug discovery, integrating functionalities for molecular visualization, modeling, and simulations. It’s leveraged by biologists and chemists for applications in pharmaceutical and biotechnological research by supporting complex tasks such as protein-ligand design and molecular simulations.
Which operating systems are compatible with MOE 2022.02?
MOE operates on Windows, Linux, Unix, and macOS, making it accessible for a wide range of users across various platforms. This cross-platform compatibility ensures flexibility within diverse research environments.
How has MOE improved in the 2022.02 version?
The 2022.02 version features enhanced user interface optimizations and additional scientific computing functionalities, including more powerful search options and improved molecular docking capabilities. These updates aim to streamline workflows and improve the accuracy of molecular simulations and analyses.







Reviews
There are no reviews yet.