
Download OpenMM 8.2 – High-Performance Molecular Dynamics Simulation Toolkit
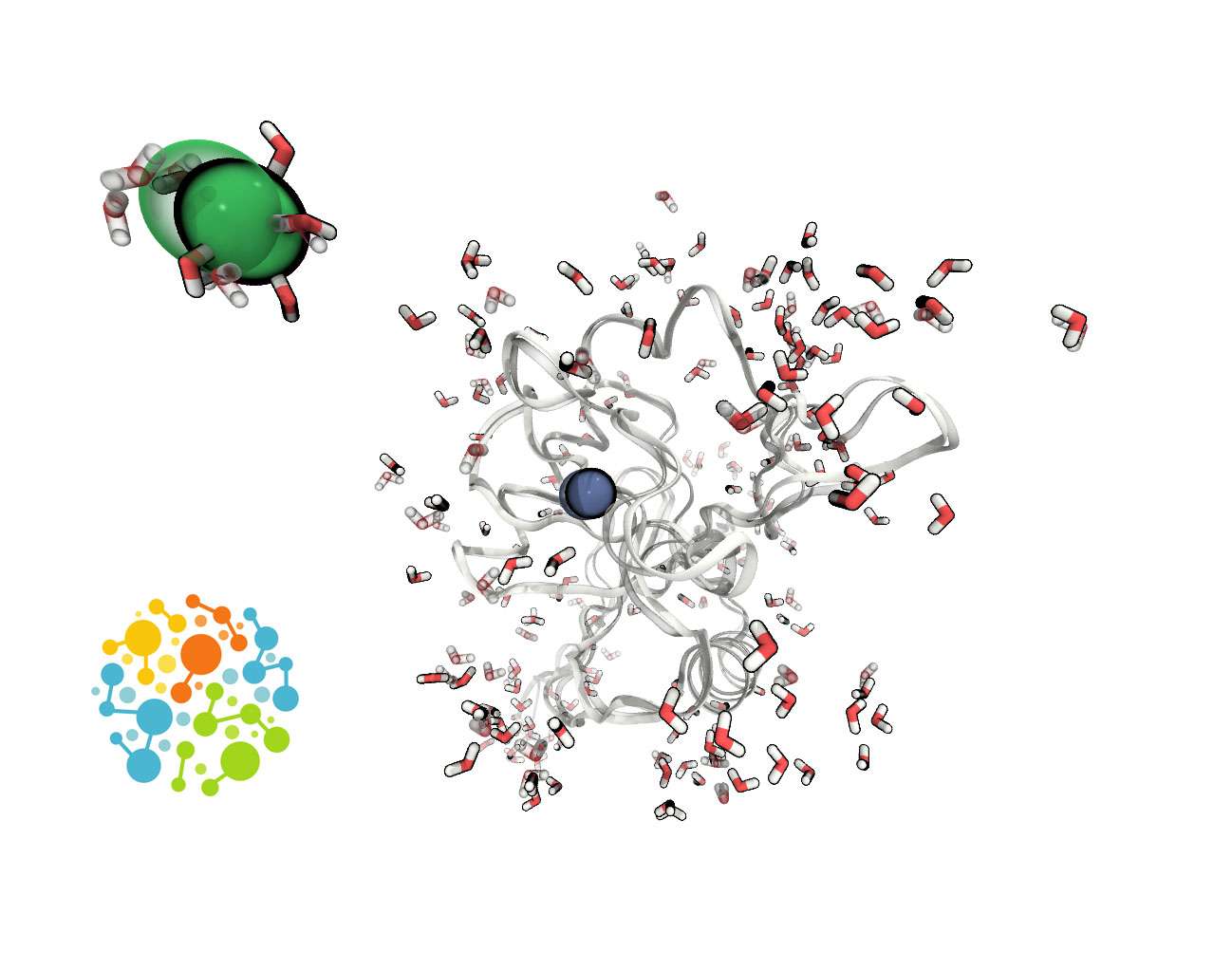
OpenMM 8.2 is an open-source molecular dynamics simulation library and toolkit, originally developed at Stanford University’s Pande lab. This high-performance software is designed for complex simulations across various scientific disciplines. It is particularly valuable for researchers in computational chemistry, biochemistry, molecular biology, pharmaceutical research, and materials science who require sophisticated tools for analyzing molecular behavior. The primary keyword for this software is OpenMM 8.2 download.
Overview of OpenMM and Its Scientific Applications
OpenMM emerges from academic research at Stanford University as a robust computational chemistry tool. Its design facilitates detailed molecular dynamics simulations, enabling scientists to investigate molecular processes at an unprecedented level of detail. The software supports a wide range of applications, from understanding the intricate folding mechanisms of proteins to modeling the interactions of drug candidates with biological targets. Professionals in pharmaceutical research, materials science, and biophysics leverage OpenMM for its scientific rigor and flexibility in exploring complex molecular systems. Its development history is rooted in academic innovation, aiming to provide powerful simulation capabilities to the scientific community.
Advanced Simulation Capabilities and Customization Options
OpenMM provides a comprehensive suite of tools for advanced molecular simulations, offering deep customization for scientific exploration. Users can select from a variety of standard force fields or implement custom ones to accurately model specific molecular interactions. The toolkit also supports diverse integration algorithms, including explicit solvent models and thermostats like Langevin, Verlet, and Nosé–Hoover, allowing researchers to tailor simulation parameters to their specific experimental conditions. This flexibility extends to both high-level users who can run standalone simulations and developers who can integrate OpenMM’s capabilities into their own custom-coded applications and workflows.
GPU Optimization and Integration in Distributed Computing
A key strength of OpenMM is its advanced GPU optimization, providing significant performance enhancements for molecular dynamics simulations. By leveraging technologies such as CUDA for NVIDIA GPUs and OpenCL for broader compatibility including AMD GPUs, OpenMM significantly accelerates computational tasks. This capability is critically important for projects like Folding@home, where OpenMM serves as a core simulation engine. Its integration into distributed computing platforms allows for the execution of massive-scale protein dynamics simulations, utilizing the collective power of networked modern hardware to tackle complex biological questions.
Compatibility and Linux Support in Version 8.2
Version 8.2 of OpenMM brings important enhancements and stability improvements, particularly for users operating on Linux platforms. This release addresses specific issues related to OpenCL and GLIBC, ensuring a more robust and reliable simulation experience. For researchers who rely on Linux for their high-performance computing environments, these updates are crucial for conducting lengthy and demanding simulations with confidence. The focus on stability in OpenMM 8.2 makes it a dependable choice for production-level research that requires consistent and accurate results.
Practical Use Cases and Industry Applications
OpenMM 8.2 finds application in numerous critical areas within scientific research and industry. Its capabilities are instrumental in detailed protein folding studies, helping to unravel the mechanisms behind diseases and develop new therapeutic strategies. In drug discovery, it simulates molecular interactions to identify promising drug candidates and optimize their efficacy. Furthermore, materials scientists utilize OpenMM to design and understand novel materials at the molecular level, predicting their properties and behaviors. The widespread use of OpenMM in projects like Folding@home demonstrates its utility in tackling large-scale scientific challenges.
Comparisons with Other Molecular Simulation Tools
Compared to other widely used molecular dynamics simulation software such as GROMACS and AMBER, OpenMM 8.2 distinguishes itself through its exceptional flexibility and GPU performance. While these tools offer powerful simulation capabilities, OpenMM’s architecture as both a library and a standalone application makes it highly adaptable for custom workflows and integrations. Its robust support for heterogeneous computing environments, leveraging both CUDA and OpenCL, allows for efficient execution on a diverse range of modern hardware, often providing a competitive edge in simulation speed and scalability for specific computational chemistry tasks.
Frequently Asked Questions
What are the primary advantages of using OpenMM 8.2 for molecular dynamics simulations?
OpenMM 8.2 offers flexible and high-performance molecular dynamics simulations, especially optimized for GPUs via CUDA and OpenCL. It allows users to customize force fields and integrators while also providing built-in options, catering to both developers and high-level users.
Can OpenMM 8.2 run on Linux platforms and utilize GPUs?
Yes, OpenMM 8.2 supports Linux and is optimized for GPU acceleration on both AMD and NVIDIA GPUs using OpenCL and CUDA, making it suitable for high-performance computing environments common in research.
How is OpenMM 8.2 used in the Folding@home project?
OpenMM 8.2 serves as the molecular dynamics engine in Folding@home’s GPU cores (specifically cores 26 and 27), enabling large-scale protein dynamics simulations by leveraging distributed GPU computing resources worldwide.







Reviews
There are no reviews yet.